Capítulo 3
Diagnóstico genético
¿Qué protocolo es recomendable para el estudio genético de distrofia muscular de Duchenne/distrofia muscular de Becker?
P. Gallano Petit
Las distrofinopatías son enfermedades ligadas al cromosoma X, incluida la distrofia muscular de Duchenne (DMD) y la distrofia muscular de Becker (DMB), debido a mutaciones del gen DMD. En los últimos años, la aplicación de nuevas tecnologías ha influido en las pruebas genéticas diagnósticas de las distrofinopatías.
Las deleciones o duplicaciones de uno o más exones son el tipo de mutaciones predominantes en la patología molecular del gen DMD (~ 70%). Por consiguiente, la primera prueba que realizar en el diagnóstico genético sería el análisis de este número de copias exónicas (Fig. 1)1.
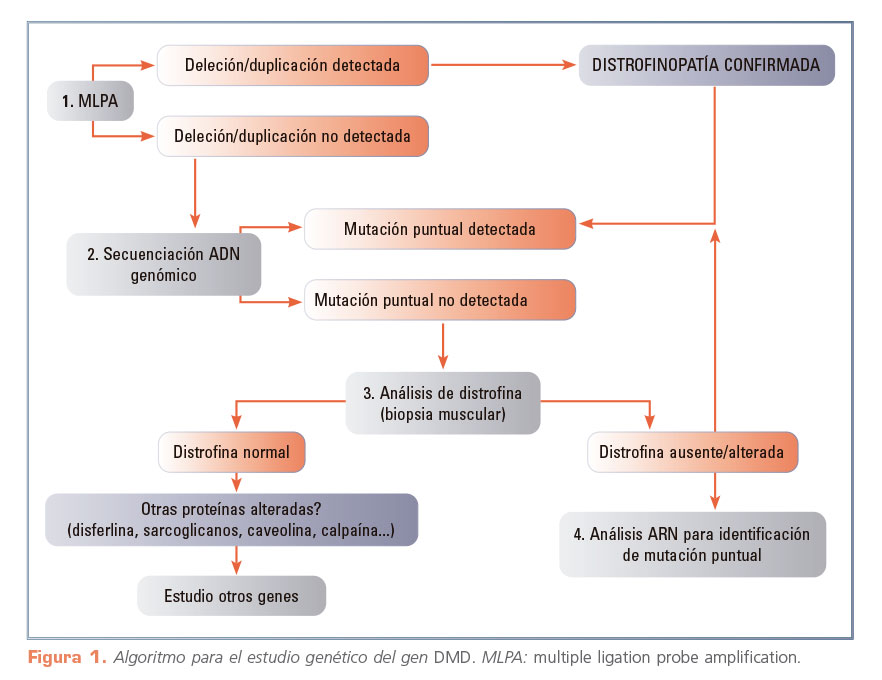
Entre los muchos métodos cuantitativos disponibles, la MLPA (multiple ligation probe amplification) es actualmente el más utilizado debido a su alta sensibilidad y reducido coste económico2. En caso de identificarse la deleción de un único exón, es obligado confirmar el resultado mediante otro método diferente (p. ej., secuenciación Sanger del exón en cuestión) y descartar que se trata de un falso positivo originado por un cambio puntual (polimórfico o patogénico) en la secuencia del ADN del paciente que impide el correcto funcionamiento de la técnica de MLPA3.
Las mutaciones responsables del 30% restante de los pacientes consisten en mutaciones puntuales (nonsense, frameshift, pequeñas indel). Dado que las mutaciones puntuales en el gen DMD son muy variadas y se distribuyen a lo largo del gen, se deben analizar los 79 exones y las secuencias intrónicas flanqueantes. Esto se realiza, generalmente, mediante secuenciación del ADN genómico del paciente. Si bien es un método preciso, la secuenciación Sanger requiere mucho tiempo, dado el gran tamaño del gen DMD4, por lo que hoy se ve sustituida por la secuenciación masiva o NGS (secuenciación de nueva generación), capaz de dar un resultado en un espacio de tiempo más corto5. Si el resultado fuera negativo, se procedería en última instancia a secuenciar el ADNc derivado de ARN obtenido de biopsia muscular, lo que permitiría identificar la presencia de una mutación puntual.
¿Es importante el estudio genético de portadoras de DMD?
L. González Quereda
El gen DMD está localizado en el brazo corto del cromosoma X (Xp21)6,7 y se transmite de forma recesiva ligado al cromosoma X. Dado que los varones tienen un único cromosoma X y que las mujeres poseen dos, si un varón hereda el cromosoma X con la mutación en el gen DMD desarrollará la enfermedad, mientras que las mujeres serán portadoras, en su gran mayoría asintomáticas. Una mujer portadora presenta una probabilidad del 50% de transmitir el alelo mutado en cada gestación (independientemente del sexo del feto). No obstante, debe tenerse en cuenta que se estima que alrededor de un 30% de los casos de distrofinopatías están causados por mutaciones de novo8, es decir, mutaciones que aparecen por primera vez en la familia y que no están presentes en generaciones anteriores. Ante el primer caso de DMD en una familia, y una vez identificada la mutación responsable de la patología en el caso índice, es fundamental ofrecer un estudio genético a la madre del paciente para verificar si es portadora de la enfermedad. La información que resulte de su test genético será fundamental para el asesoramiento genético de los demás miembros de la familia. Si la mujer resulta ser portadora, podrá ser asesorada genéticamente y conocer el riesgo de recurrencia en su descendencia, la probabilidad de volver a transmitir la enfermedad en futuros embarazos y las diferentes opciones reproductivas disponibles. La información acerca del status de portadora será también importante para otras mujeres de la familia por vía materna (hermanas, tías…), que a su vez también podrían ser portadoras de la mutación en DMD.
Si el resultado del test genético revela que la mujer es no portadora, la probabilidad de transmitir la enfermedad a futuras generaciones disminuye considerablemente, aunque no exime de cierto riesgo, dado que existe una probabilidad de alrededor de un 7-10% de que la mujer sea portadora de un mosaicismo germinal9. En este caso la mutación no se detectará en el ADN genómico procedente de sangre periférica, ya que la mutación solo estará presente en parte de sus células germinales.
Conocer la condición de portadora también ofrece a la mujer la posibilidad de recibir un seguimiento médico, con particular interés en el aspecto cardiológico, para de este modo detectar de forma precoz afectaciones cardiológicas que en ocasiones se presentan en las mujeres portadoras10.
¿Cuál es la importancia de una adecuada interpretación del estudio genético y su implicación en opciones terapéuticas?
P. Gallano Petit
El gen DMD está constituido por 79 exones (regiones codificadoras) a lo largo de alrededor de 2.200 kb (~ 2 millones de nucleótidos) y codifica para la proteína denominada distrofina, cuya isoforma muscular consta de 3.654 aminoácidos (aa). El código de lectura de la distrofina se lleva a cabo en los ribosomas, los cuales traducen el mensaje a modo de tripletes o codones gracias al ARN mensajero que es el ácido nucleico intermediario o «portavoz» del ADN genómico. El código genético determina que cada aa que se integre en la cadena proteica vendrá codificado por tres nucleótidos.
El número de nucleótidos que constituye cada exón no siempre es múltiplo de tres. A menudo el exón finaliza con un único nucleótido al que le esperan dos nucleótidos en el inicio del siguiente exón, para de este modo entre los tres incorporar un nuevo aa. De modo opuesto, si la secuencia exónica finaliza con dos nucleótidos, estos se traducirán junto con el primer nucleótido del siguiente exón (Fig. 2)11,12.

Una de las aproximaciones terapéuticas de la DMD es la terapia de salto de exón13. Este tipo de terapia tiene como objetivo restaurar el código de lectura de la distrofina de los pacientes con Duchenne que se ha visto interrumpido por la presencia de una deleción o una mutación puntual, las cuales han originado la aparición de uno de los tres codones stop del código genético (UAA, UAG UGA). Así, por ejemplo, el salto del exón 51 (Fig. 2) restaura la deleción del exón 50 conectando el exón 49 al exón 52.
Otra aproximación terapéutica es la translectura (readthrough) mediante el fármaco PTC124, que tiene como objetivo anular la lectura de codones stop prematuros producidos por mutaciones nonsense exclusivamente, es decir, aquellos cambios nucleotídicos que directamente crean uno de los tres codones stop, y respetando al mismo tiempo la lectura del codón natural de final de la traducción14.
A diferencia de las mutaciones nonsense, las mutaciones puntuales de tipo frameshift originan un desfase del molde de lectura de la proteína y secundariamente acaban originando siempre la aparición de un codón stop prematuro, a una determinada distancia del lugar donde se produjo la mutación. Este tipo de mutaciones no son reparables mediante este tipo de terapia. De ahí que en ambas aproximaciones terapéuticas es imprescindible la clara definición de la mutación identificada.
Bibliografía
1. Fratter C, Dalgleish R, Allen SK, Santos R, Abbs S, Tuffery-Giraud S, et al. EMQN best practice guidelines for genetic testing in dystrophinopathies. Eur J Hum Genet. 2020;28(9):1141-59.
2. Lalic T, Vossen RHAM, Coffa J, Schouten JP, Guc-Scekic M, Radivojevic D, et al. Deletion and duplication screening in the DMD gene using MLPA. Eur J Hum Genet. 2005;13:1231-4.
3. Santos R, Goncalves A, Oliveira J, Vieira E, Vieira JP, Evangelista T, et al. New variants, challenges and pitfalls in DMD genotyping: implications in diagnosis, prognosis and therapy. J Hum Genet. 2014;59:454-64.
4. Flanigan KM, von Niederhausern A, Dunn DM, Alder J, Mendell JR, Weiss RB. Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet. 2003;72:931-9.
5. Gonzalez-Quereda L, Rodriguez MJ, Diaz-Manera J, Alonso-Perez J, Gallardo E, Nascimento A, et al Targeted next-generation sequencing in a large cohort of genetically undiagnosed patients with neuromuscular disorders in Spain. Genes (Basel). 2020;11(5):539.
6. Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkelet LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509-17.
7. Kunkel LM, Monaco AP, Hoffman E, Koenig M, Feener C, Bertelson C. Molecular studies of progressive muscular dystrophy (Duchenne). Enzyme. 1987;38(1-4):72-5.
8. GrimmT, Kress W, Meng G, Müller CR. Risk assessment and genetic counselling in families with Duchenne muscular dystrophy. Acta Myol. 2012;31(3):179-83.
9. Bakker E, van Broeckhoven C, Bonten EJ, van de Vooren MJ, Veenema H, van Hul W, et al. Germline mosaicism and Duchenne muscular dystrophy mutations. Nature. 1987;329(6139):554-6.
10. Juan-Mateu J, Rodríguez MJ, Nascimento A, Jiménez-Mallebrera C, González-Quereda L, Rivas E, et al. Prognostic value of X-chromosome inactivation in symptomatic female Carriers of dystrophinopathy. Orphanet J Rare Dis. 2012;7:82.
11. Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2(1):90-5.
12. Aartsma-Rus A, van Deutekon JCT, Fokkema IF, van Ommen GJB, den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that conform the reading-frame rule. Muscle Nerve. 2006;34:135-44.
13. van Ommen GJ, van Deutekom J, Aartsma-Rus A. The therapeutic potential of antisense exon skipping. Curr Opin Mol Ther. 2008;10(2):140-9.
14. Campbell C, Barohn RJ, Bertini E, Chabrol B, Comi GP, Darras BT, et al. Meta-analyses of ataluren randomized controlled trials in nonsense mutation Duchenne muscular dystrophy. J Comp Eff Res. 2020;9(14):973-84.